The U.S. Food and Drug Administration fundamentally altered the trajectory of auditory medicine late Thursday, April 23, 2026, granting accelerated approval to lunsotogene parvec-cwha, a therapy that targets the molecular root of congenital hearing loss. Marketed by Regeneron Pharmaceuticals under the brand name Otarmeni, the therapy is an adeno-associated virus (AAV) vector-based treatment engineered to reverse profound sensorineural hearing loss caused by biallelic mutations in the OTOF gene.
For decades, the standard of care for children born deaf has been mechanical: surgical cochlear implants that bypass the ear’s defective biological hardware to stimulate the auditory nerve directly with electrical impulses. While effective, these implants render a metallic, often robotic approximation of sound, stripping away the nuanced acoustics required to naturally process pitch, tone, and complex musicality. Otarmeni replaces this mechanical workaround with a permanent biological restoration.
This approval marks the first time the FDA has cleared a genetic medicine for an inherited form of hearing loss, pulling a long-theorized concept out of the laboratory and into clinical practice. The decision was pushed through the agency’s speedy review program a mere 61 days after Regeneron submitted its biologics license application, underscoring the intense regulatory momentum behind neurosensory genetic therapies.
However, the headlines celebrating the arrival of this therapy obscure a highly complex web of boardroom calculations, delicate viral engineering, and regulatory maneuvers. The development of Otarmeni was nearly derailed multiple times by the physical constraints of inner-ear surgery and the biological limits of viral packaging. Understanding how this therapy finally reached the market requires looking past the FDA’s announcement and examining the microscopic, economic, and political machinery that brought it into existence.
The Biological Bottleneck: Otoferlin and the Ribbon Synapse
To understand the specific engineering behind Otarmeni, one must first examine why the OTOF gene was chosen as the inaugural target for inner-ear gene therapy.
Congenital hearing loss affects roughly 1.7 out of every 1,000 children born in the United States. The vast majority of these cases stem from structural defects where the sensory hair cells of the cochlea—the tiny, ciliated structures that convert mechanical sound wave vibrations into chemical signals—are either missing, malformed, or progressively deteriorate. Attempting to regenerate a structurally compromised cochlea requires initiating complex, multi-stage cellular growth that remains years away from human trials.
OTOF-related hearing loss presented a uniquely solvable biological bottleneck. In patients with mutations in the OTOF gene, the macro-structure of the inner ear is perfectly intact. The outer hair cells function properly, and the microscopic cilia move in sync with incoming sound vibrations. The anatomical hardware is flawless, but the biochemical software is missing a single, critical line of code.
The OTOF gene provides the cellular instructions for synthesizing otoferlin, a calcium-sensing protein expressed heavily in the inner hair cells. When a sound wave enters the ear and bends the cilia, calcium channels open, flooding the base of the hair cell with calcium ions. In a healthy ear, otoferlin acts as a specialized sensor that detects this calcium influx and immediately triggers synaptic vesicles—tiny packets filled with neurotransmitters—to fuse with the cell membrane and release their chemical payload across the ribbon synapse to the auditory nerve.
Without functional otoferlin, the vesicles cannot fuse. The physical sound wave enters the ear, the cilia dance, and the calcium floods the cell, but the signal dies at the synapse. The brain receives nothing. The auditory nerve waits in absolute silence.
Because the structural architecture of the ear remains preserved in these patients, scientists realized that if they could successfully smuggle a working copy of the OTOF gene into the existing, living inner hair cells, the cells could theoretical begin producing otoferlin and the biological switch would be flipped. The ear would suddenly come online.
Overcoming the Viral Packaging Limit
The theory was sound, but the mechanics of delivering the gene were intensely problematic. The development of a functional genetic deafness cure has historically been stalled by the sheer physical size of the required biological payload.
Like most gene therapies, Regeneron’s Otarmeni (originally developed under the clinical designation DB-OTO) relies on a modified, non-pathogenic adeno-associated virus (AAV) to act as a delivery vehicle. AAVs are favored in gene therapy because they do not integrate their payload directly into the host genome, which reduces the risk of triggering oncogenic (cancer-causing) mutations. Instead, they deposit their genetic cargo into the cell nucleus, where it exists as an episome—an independent circular piece of DNA that the cell can read and transcribe.
The fatal flaw of AAVs, however, is their strict packaging capacity. An AAV capsid can physically hold only about 4.7 kilobases (kb) of genetic material. The full-length human OTOF gene is notoriously massive, clocking in at nearly 6 kb. It physically cannot fit inside a standard AAV vector.
Competitors in the space approached this hurdle through fragmentation. Eli Lilly and its subsidiary Akouos, which are developing a rival therapy known as AK-OTOF, engineered a dual-vector system. They split the OTOF gene into two distinct halves, packaged each half into a separate virus, and relied on the viruses co-infecting the exact same inner hair cell. Once inside, the two halves of the genetic sequence recombine to form a complete, functional gene. While ingenious, dual-vector systems are inherently less efficient; if a cell only absorbs one of the two viruses, the therapy fails in that specific cell.
Regeneron, which acquired the foundational technology for Otarmeni through its $109 million buyout of Decibel Therapeutics in 2023, took a highly proprietary approach to the vector architecture. To circumvent the size limits and maximize transcription efficiency, Regeneron's scientists utilized a highly selective delivery sequence. The critical breakthrough was not just the viral capsid, but the promoter—the genetic "on-switch" that tells the cell to read the newly delivered DNA.
Otarmeni utilizes a proprietary, cell-specific Myo15 promoter. This promoter is biologically tuned to activate only in native hair-cell compartments. If the viral vector inadvertently infects supporting cells, glial cells, or surrounding tissue in the cochlea, the DNA payload remains dormant. By restricting expression exclusively to the hair cells that normally produce otoferlin, Regeneron effectively eliminated off-target toxicity, allowing them to use a highly concentrated viral titer without triggering a destructive immune cascade in the delicate, fluid-filled compartments of the ear.
The Surgical Reality: Navigating the Round Window
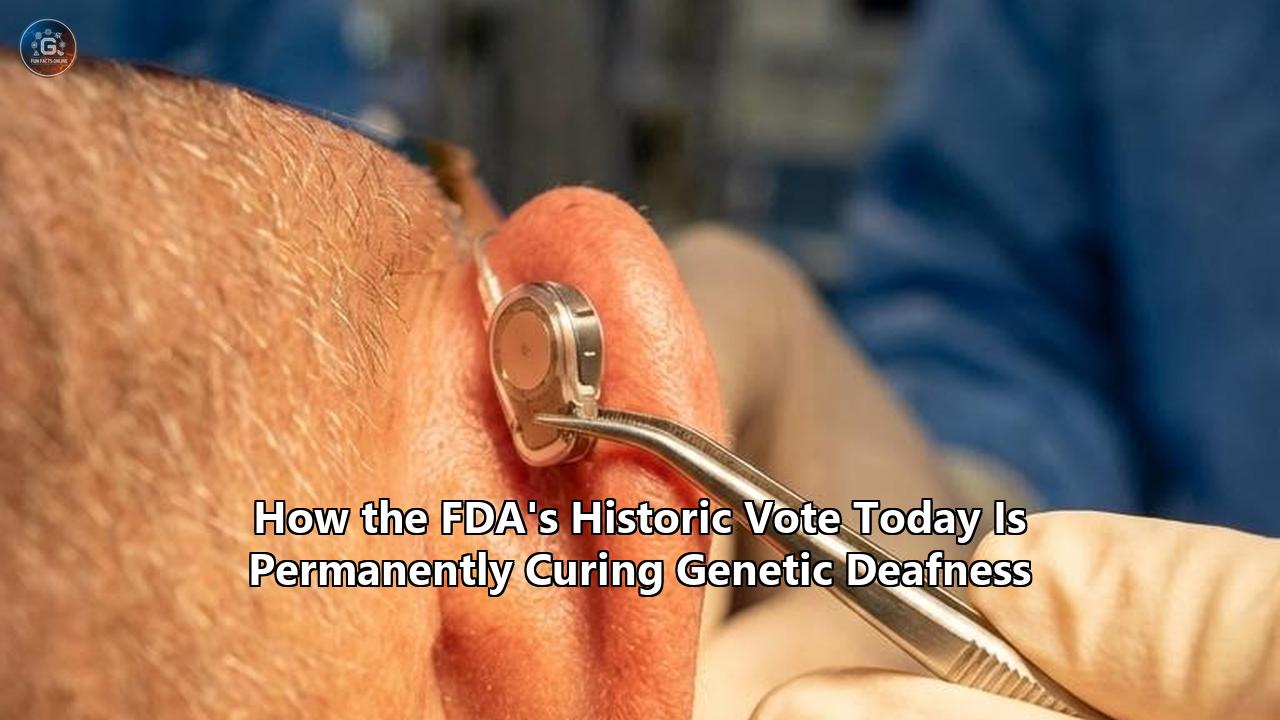
Delivering the therapy presented a secondary, equally daunting challenge. The inner ear is a closed, heavily fortified system embedded deep within the temporal bone, one of the densest bones in the human body. It is filled with a specialized fluid called perilymph, and any significant alteration in fluid pressure or introduction of foreign biological matter can trigger inflammation that permanently destroys whatever residual auditory function the patient might have.
“No one has ever tried to use a viral vector to target the cells of the inner ear before. No one has done this surgery before for humans,” noted Dr. A. Eliot Shearer, a pediatric otolaryngologist at Boston Children’s Hospital, Harvard Medical School associate professor, and a key investigator on the clinical trials. The physical act of administration required adapting techniques previously reserved for cochlear implantation.
Under general anesthesia, surgeons access the cochlea by gently lifting the eardrum and navigating through the middle ear to locate the round window membrane—a microscopic, tissue-thin barrier separating the middle ear from the fluid-filled inner ear. A specialized micro-catheter is carefully inserted through this membrane. The Otarmeni suspension is then pushed into the perilymph fluid over several minutes.
The margin for error is razor-thin. Inject the fluid too quickly, and the hydraulic pressure will shear the delicate hair cells off the basilar membrane, causing permanent mechanical deafness. Inject it too slowly, and the viral vector fails to disseminate evenly throughout the spiraling chambers of the cochlea, leaving certain frequency ranges dead while restoring others.
The successful translation of this surgical protocol from preclinical models to human pediatric patients is arguably as significant an achievement as the molecular biology of the drug itself. It proves that the cochlea can endure direct viral inoculation without succumbing to immediate inflammatory rejection.
The CHORD Trial: The Data Behind the Decision
The accelerated FDA approval was anchored by data from the Phase 1/2 CHORD study, an ongoing, open-label, single-arm trial that enrolled 20 individuals ranging in age from 10 months to 16 years.
The trial was designed with aggressive primary endpoints. Patients were required to have no prior cochlear implant in the ear slated for treatment, as the physical insertion of a mechanical implant destroys the biological structures required for the gene therapy to work. Efficacy was measured primarily through pure tone audiometry (PTA) assessments and auditory brainstem responses (ABR).
The data presented to the FDA review committee was unusually definitive for a Phase 1/2 study, a factor that heavily influenced the rapid 61-day turnaround. At the 24-week post-infusion mark, 16 of the 20 patients (80%) achieved hearing improvements passing the critical threshold of ≤70 decibel hearing level (dB HL). This ≤70 dB HL mark is the clinical standard for natural, conversational hearing where a cochlear implant is no longer deemed medically necessary.
Even more compelling were the secondary objective measures. Because the subjects included infants unable to consciously indicate whether they heard a sound, researchers relied on ABR testing—placing electrodes on the scalp to measure the exact electrical signals traveling from the auditory nerve to the brainstem in response to an acoustic click.
Before treatment, the children in the CHORD trial registered zero electrical brainstem activity even at sounds exceeding 90 decibels (the equivalent of a lawnmower running directly next to the ear). By 24 weeks post-treatment, 14 of the participants (70%) demonstrated robust auditory brainstem responses at ≤90 dB. Among a subset of the earliest treated patients, the results were more profound: several children achieved normal or near-normal acoustic hearing across all frequency ranges.
The qualitative data provided the emotional leverage needed to push the therapy into the public consciousness. Regeneron documented the case of Travis Smith, a two-and-a-half-year-old born with profound OTOF-related deafness. Prior to receiving DB-OTO, he was entirely unresponsive to sound. Months after his unilateral infusion, clinical observers recorded him actively responding to music and localized voices.
“My son was 100% deaf,” his mother Sierra Smith stated during the trial follow-ups. “Didn't say mama, didn't even know he had a name... He loves playing his little guitar. He loves listening to Bruno Mars”.
While these individual case studies are powerful, the FDA review committee focused heavily on the stability of the intervention. The durability of a single-dose genetic deafness cure is the primary lingering question. Otarmeni operates via an episomal DNA vector; because the new OTOF gene does not integrate into the host cell's native chromosomes, there is a theoretical risk that expression could wane over years or decades. Since hair cells do not actively divide and regenerate in humans, the episome should remain stable inside the cell nucleus indefinitely, but clinical confirmation of this long-term stability will require years of continued post-market surveillance.
The Economics of a "Free" Drug and Priority Vouchers
One of the most heavily scrutinized aspects of Thursday's FDA announcement was Regeneron’s unprecedented pricing strategy. The company declared it will provide Otarmeni entirely free of charge to all eligible patients in the United States (though hospitals may still charge for the surgical administration).
On its face, giving away a novel, highly complex biologic seems financially counterintuitive for a publicly traded biopharmaceutical giant. Gene therapies typically launch with multi-million dollar price tags; CSL Behring's Hemgenix for hemophilia B costs $3.5 million per dose, and Bluebird Bio's Zynteglo for beta-thalassemia runs $2.8 million.
But offering a genetic deafness cure at no cost is not merely an act of corporate philanthropy; it is a calculated regulatory and economic maneuver intrinsically tied to the mechanisms of the FDA.
Otarmeni was approved under the FDA’s Commissioner’s National Priority Voucher (CNPV) program, a highly selective pathway designed to incentivize the development of drugs for ultra-rare pediatric diseases. OTOF-related hearing loss is exceptionally rare; it is diagnosed in roughly 50 newborns per year in the United States, and affects perhaps 200,000 people globally. Even at a price point of $2 million per dose, the total addressable market in the U.S. is too small to quickly recoup the $109 million Regeneron spent acquiring Decibel Therapeutics, let alone the costs of clinical trials and proprietary AAV manufacturing.
However, by successfully pushing Otarmeni across the regulatory finish line, Regeneron was awarded a rare pediatric disease priority review voucher. This voucher is a transferable regulatory golden ticket. It allows the holder to force the FDA to review any future new drug application in six months rather than the standard ten months.
In the high-stakes pharmaceutical industry, cutting four months off the review time for a potential blockbuster drug—such as a mass-market obesity injectable or a next-generation oncology biologic—can translate to hundreds of millions, if not billions, of dollars in early revenue before generic competition enters the market. Furthermore, these vouchers are legally tradable. In recent years, companies have sold priority review vouchers to other pharmaceutical firms on the open market for upwards of $100 million to $110 million each.
By distributing Otarmeni for free, Regeneron achieves a multifaceted victory. They completely bypass the grueling, multi-year process of negotiating coverage with private health insurers and state Medicaid programs, which frequently balk at high upfront gene therapy costs. They guarantee immediate market penetration and clinical adoption, boxing out impending competitors. They harvest invaluable, real-world, long-term human data on inner-ear viral vector delivery to inform their future pipeline. And they walk away with a priority review voucher whose financial value likely exceeds the direct commercial potential of the drug itself.
“We are honored to be in the position to be the first company to ever offer such a gene therapy advance for free to those in the U.S.,” stated Dr. George Yancopoulos, Regeneron’s president and chief scientific officer, framing the move as a demonstration that the biopharmaceutical industry "can be a genuine force for good in the world". Behind the public relations triumph, the financial architecture of the deal perfectly illustrates how modern pharmaceutical development leverages regulatory loopholes to fund niche genetic cures.
The Global Arms Race and Competitive Pressures
Regeneron may have crossed the finish line first, but the OTOF gene space is highly crowded, reflecting a massive influx of capital into auditory neuroscience.
Eli Lilly’s AK-OTOF is moving aggressively through its own Phase 1/2 trial (AK-OTOF-101). Early data released at medical conferences throughout 2024 and 2025 indicated that AK-OTOF achieved rapid pharmacologic hearing restoration, with an 11-year-old participant showing restored thresholds across all tested frequencies within 30 days of administration. Lilly’s dual-vector system has shown proof-of-concept, and the company is expected to leverage its massive manufacturing footprint to push for an approval before their trial’s estimated completion in late 2028.
Meanwhile, European biotechnology firm Sensorion has been steadily advancing its AAV vector-based therapy, SENS-501 (also known as OTOF-GT), in the Phase 1/2 AUDIOGENE clinical trial. In early 2025, an independent Data Monitoring Committee cleared Sensorion to escalate to a higher dose cohort after reviewing favorable safety data from infants aged 6 to 31 months.
The most intense early pressure, however, came from parallel research in China. In mid-2024, researchers from Fudan University and Refreshgene Therapeutics published data regarding their own AAV1-hOTOF gene therapy, demonstrating robust hearing improvement over 26 weeks in multiple children. The international race forced the FDA to aggressively utilize its accelerated approval pathways to ensure a U.S.-developed therapy reached the market concurrently with global advancements.
Regeneron’s approval essentially resets the regulatory floor. With Otarmeni now established as an approved biological option, clinical trials for mechanical cochlear implants in OTOF-mutated patients will face severe ethical hurdles. Enrolling a child in a trial for a mechanical device when a free, FDA-approved biological cure exists will be highly scrutinized by institutional review boards.
Expanding the Genetic Toolbox: Beyond OTOF
For Regeneron, Otarmeni is merely the vanguard of a much broader strategic shift. The 50 infants born annually in the U.S. with OTOF mutations represent a fraction of a percent of the total population suffering from inherited hearing loss.
“Otarmeni is really our first foray into genetic medicine,” Aris Baras, senior vice president and general manager at the Regeneron Genetics Center, detailed in industry briefings prior to the approval. “We've always had this dream, this vision, that we could do a lot in a space like hearing loss. It shows that we can do something that we weren't sure was possible: restore kind of complete loss of hearing to normal hearing”.
The company is heavily investing in the biology of acquired and complex structural forms of deafness. While a single-gene replacement works for otoferlin deficiencies, treating the other 1 in 500 infants born with hearing loss requires a more diversified approach.
The next frontier involves targeting structural mutations, such as variants in the GJB2 gene (which produces the Connexin 26 protein, responsible for maintaining potassium levels in the cochlea) and the complex polygenic pathways involved in Usher Syndrome, which causes both deafness and progressive blindness.
Because structural proteins require incredibly precise cellular regulation, a brute-force AAV vector delivering a continuously active gene might cause overexpression and further cellular damage. To address this, Regeneron and its competitors are expanding their modalities beyond standard viral delivery. The company has confirmed active development of small interfering RNA (siRNA) therapeutics and specialized monoclonal antibodies designed to modulate, rather than strictly replace, genetic expression within the inner ear.
The surgical protocols validated by Otarmeni’s approval provide the physical delivery mechanism for these future therapies. By proving that the cochlea can safely tolerate intracochlear infusions without destroying residual hearing, surgeons have established a viable pharmacokinetic pathway to the inner ear.
The Post-Market Requirements and Unresolved Questions
Because Otarmeni was pushed through via the accelerated approval pathway, the FDA’s green light is provisional. Regeneron is legally bound to provide ongoing post-marketing data to retain its commercial authorization.
The FDA mandate requires Regeneron to verify two critical endpoints: long-term durability of the hearing improvement, and verifiable secondary effects on real-world speech development and quality of life. Restoring the biological ability to detect sound is only the first half of the equation; neuroplasticity dictates the rest.
In children born deaf, the auditory cortex of the brain—deprived of sensory input—rapidly begins to rewire itself to process visual or tactile information. If intervention is delayed too long, the brain loses the capacity to interpret complex acoustic data, even if the ear suddenly begins transmitting perfect signals. The CHORD trial targeted a wide age range (10 months to 16 years) specifically to gather data on this neuroplastic window.
Early results show that early intervention is heavily correlated with better outcomes for organic speech development. Children treated at 11 months, like the British patient Opal Sandy who received a similar experimental therapy, demonstrate a rapid integration of acoustic stimulus, catching up to their hearing peers in vocal milestones. For the older adolescents in the CHORD trial, tracking how effectively the adult brain can learn to decode newly acquired biological hearing will define the therapeutic limits of the intervention.
The timeline for the next genetic deafness cure targeting different mutations depends heavily on these early post-market datasets. If the episomal OTOF expression proves stable at the three- and five-year marks without triggering delayed-onset inflammation, regulatory confidence will surge for more complex inner-ear targets.
For now, the era of treating genetic deafness solely as a mechanical deficit has ended. The biological hardware of the cochlea has proven accessible, and the genetic software can be rewritten. The focus now shifts from proving the science is possible to scaling its reach, turning a niche regulatory triumph into a broad, multi-target platform for auditory restoration.
Reference:
- https://medcitynews.com/2026/04/regeneron-gene-therapy-hearing-loss-otarmeni-fda-approval-otof-otoferlin-db-oto-regn/
- https://www.pharmacytimes.com/view/fda-grants-accelerated-approval-to-otarmeni-first-and-only-gene-therapy-for-genetic-hearing-loss
- https://www.2minutemedicine.com/regeneron-db-oto-gene-therapy-restores-genetic-hearing-loss/
- https://www.contemporarypediatrics.com/view/fda-approves-first-gene-therapy-for-genetic-hearing-loss
- https://www.biospace.com/fda/fda-approval-of-regenerons-hearing-loss-gene-therapy-breaks-barriers
- https://www.cgtlive.com/view/world-hearing-day-2025-looking-back-progress-gene-therapy
- https://www.bioworld.com/articles/730604-a-free-gene-therapy-regenerons-otarmeni-approved-for-hearing-loss?v=preview
- https://www.fiercepharma.com/pharma/regeneron-ushers-new-genetic-medicine-era-groundbreaking-gene-therapy-approval
- https://firstwordpharma.com/story/7205633
- https://www.clinicaltrialsarena.com/news/akouos-gene-therapy-trial/
- https://clinicaltrials.gov/study/NCT05821959
- https://hearingpractitionernews.com.au/gene-therapy-restores-hearing-in-baby-and-children-with-genetic-deafness/